A blue-print for genome diagnostics for patients with rare diseases in Germany (genomDE).
Abstract
Most individuals with rare diseases initially consult their primary care physician. For a subset of rare diseases, efficient diagnostic pathways are available. However, ultra-rare diseases often require both expert clinical knowledge and comprehensive genetic diagnostics, which poses structural challenges for public healthcare systems. To address these challenges within Germany, a novel structured diagnostic concept, based on multidisciplinary expertise at established university hospital centers for rare diseases (CRDs), was evaluated in the three year prospective study TRANSLATE NAMSE. A key goal of TRANSLATE NAMSE was to assess the clinical value of exome sequencing (ES) in the ultra-rare disease population. The aims of the present study were to perform a systematic investigation of the phenotypic and molecular genetic data of TRANSLATE NAMSE patients who had undergone ES in order to determine the yield of both ultra-rare diagnoses and novel gene-disease associations; and determine whether the complementary use of machine learning and artificial intelligence (AI) tools improved diagnostic effectiveness and efficiency. ES was performed for 1,577 patients (268 adult and 1,309 pediatric). Molecular genetic diagnoses were established in 499 patients (74 adult and 425 pediatric). A total of 370 distinct molecular genetic causes were established. The majority of these concerned known disorders, most of which were ultra-rare. During the diagnostic process, 34 novel and 23 candidate genotype-phenotype associations were delineated, mainly in individuals with neurodevelopmental disorders. To determine the likelihood that ES will lead to a molecular diagnosis in a given patient, based on the respective clinical features only, we developed a statistical framework called YieldPred. The genetic data of a subcohort of 224 individuals that also gave consent to the computer-assisted analysis of their facial images were processed with the AI tool Prioritization of Exome Data by Image Analysis (PEDIA) and showed superior performance in variant prioritization. The present analyses demonstrated that the novel structured diagnostic concept facilitated the identification of ultra-rare genetic disorders and novel gene-disease associations on a national level and that the machine learning and AI tools improved diagnostic effectiveness and efficiency for ultra-rare genetic disorders.
What is the Selektivvertrag (§140 SGBV) “Exom-Sequenzierung bei Seltenen Erkrankungen”?
Since the evaluation of TRANSLATE NAMSE was positive, the concept is continued as a Selektivvertrag. That basically means the parties that can participate are paid a little bit more money for Wissensgenerierende Krankenversorgung, that is they continue with research until a case is solved. A crucial part of solving a case that is due to a novel disease (otherwise it would have been simple to diagnose it), is sharing data. The members of the Selektivvertrag share variant of unknown clinical significance (VUCS) and clinical features of a patient, e.g. intellectual disability, in HPO terminology, in order to identify similar cases. A new monogenic disorder requires at least three cases with similar mutations e.g. in the same gene, and similar phenotype, e.g. a facial gestalt that’s alike. This is first done on the level of the members of the Selektivvertrag (national level) and if this is not sufficient on a global level via the Match Maker Exchange network. If your IP address is already on the whitelist, you can reach Exome AG server here:
If you are a member of the Selektivvertrag and would like to be onboarded, please contact pkrawitz@uni-bonn.de and schmida@uni-bonn.de, or call +49 171 7889198.
Can an exome solve your case? YieldPred knows!
The diagnostic yield is highly dependent on the disease group, the clinical features, and the genetic test. Based on the TRANSLATE NAMSE cohort the tool YieldPred was developed to predict the diagnostic yield that can be achieved with exome sequencing (ES). YieldPred is based on least absolute shrinkage and selection operator (LASSO), which is a multi-regression analysis that we performed on the HPO annotations of all cases. Users can specify the age, sex, and assigned HPO terms of their patient while the remaining confounders are estimated as the mean effect of the TRANSLATE NAMSE cohort. The service provides a point estimate of the diagnostic yield as well as a relation to the densities of the diagnostic yields of TRANSLATE NAMSE patients resulting from the model.
The TRANSLATE NAMSE ES cohort (n=1,577) was randomly divided into a training set incorporating 1,256 cases (399 solved, 32%) and a test set incorporating 321 cases (99 solved, 31 %). For each of the 49 phenotypic groups (cf. clinical and laboratory phenotype data), we defined a binary predictor being 1 if the patient was assigned at least one HPO term of the respective group and 0 otherwise. The binary status of a case (1=solved, 0=not solved) was regressed on those 49 phenotypic predictors using LASSO for binary outcomes with the logit function as a link function (R package glmnet, version 4.1-4) and controlling for age (adult/child), sex (male/female), sequencing laboratory, and the use of the PEDIA workflow. Variable selection was applied only on the 49 phenotypic groups. The model was fitted on the training data and the penalty parameter was tuned via ten-fold cross-validation. The resulting model was then applied to the test set, and its predictive performance was evaluated using the Receiver Operator Characteristics (ROC) curve. The model was then refitted on the whole ES cohort of 1,577 cases and made the results available in the predictor of diagnostic yield web application.
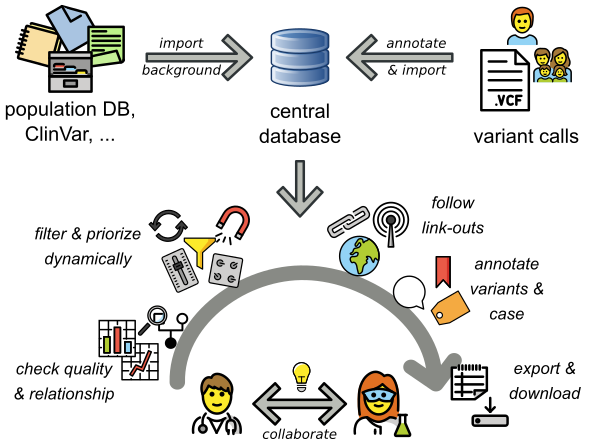
What is the most efficient way to analyze your variants? Using PEDIA!
PEDIA is a workflow that adds the results of computer-assisted evaluation of a portrait to the interpretation of variants (Prioritization of Exome Data by Image Analysis). The PEDIA approach showed superior performance compared to standard workflows that only work with molecular- and feature-based scores. The Image analysis is done by the AI GestaltMatcher, which is open-source and provided by AGD e.V.. PEDIA can be integrated in frameworks for variant analysis such as e.g. VarFish or GeneTalk, which are user-friendly web applications for the quality control, filtering, prioritization, analysis, and user-based annotation of DNA variant data with a focus on rare disease genetics. VarFish was originally developed in Berlin by CUBI (Core Unit Bioinformatics, BIH) together with the Institute of Medical and Human Genetics, Charite Berlin. Currently, a growing number of genetics institutes and TRANSLATE NAMSE members are is using VarFish and are working on extensions and improvements. You can find out more on the CUBI Website.
Code availability and Software Demos
- TNAMSE Code Repository
- PEDIA is provided as a Webservice by GeneTalk. If you are interested in a customized integration of the PEDIA protocol into your diagnostics workflow, please contact info@gene-talk.de.
- VarFish Demo shows the features of the VarFish software including user-based commenting of variants. This is the “classic” mode where data is uploaded using a back-end API by computational staff.
- VarFish Kiosk allows users to upload their own cases as VCF files and perform an analysis.
Publications
Axel Schmidt, Magdalena Danyel, Kathrin Grundmann, Theresa Brunet, Hannah Klinkhammer, Tzung-Chien Hsieh, Hartmut Engels, Sophia Peters, Alexej Knaus, Shahida Moosa, Luisa Averdunk, Felix Boschann, Henrike Sczakiel, Sarina Schwartzmann, Martin Atta Mensah, Jean Tori Pantel, Manuel Holtgrewe, Annemarie Bösch, Claudia Weiß, Natalie Weinhold, Aude-Annick Suter, Corinna Stoltenburg, Julia Neugebauer, Tillmann Kallinich, Angela M. Kaindl, Susanne Holzhauer, Christoph Bührer, Philip Bufler, Uwe Kornak, Claus-Eric Ott, Markus Schülke, Hoa Huu Phuc Nguyen, Sabine Hoffjan, Corinna Grasemann, Tobias Rothoeft, Folke Brinkmann, Nora Matar, Sugirthan Sivalingam, Claudia Perne, Elisabeth Mangold, Martina Kreiss, Kirsten Cremer, Regina C. Betz, Tim Bender, Martin Mücke, Lorenz Grigull, Thomas Klockgether, Spier Isabel, Heimbach André, Bender Tim, Fabian Brand, Christiane Stieber, Alexandra Marzena Morawiec, Pantelis Karakostas, Valentin S. Schäfer, Sarah Bernsen, Patrick Weydt, Sergio Castro-Gomez, Ahmad Aziz, Marcus Grobe-Einsler, Okka Kimmich, Xenia Kobeleva, Demet Önder, Hellen Lesmann, Sheetal Kumar, Pawel Tacik, Min Ae Lee-Kirsch, Reinhard Berner, Catharina Schuetz, Julia Körholz, Tanita Kretschmer, Nataliya Di Donato, Evelin Schröck, André Heinen, Ulrike Reuner, Amalia-Mihaela Hanßke, Frank J. Kaiser, Eva Manka, Martin Munteanu, Alma Kuechler, Kiewert Cordula, Raphael Hirtz, Elena Schlapakow, Christian Schlein, Jasmin Lisfeld, Christian Kubisch, Theresia Herget, Maja Hempel, Christina Weiler-Normann, Kurt Ullrich, Christoph Schramm, Cornelia Rudolph, Franziska Rillig, Maximilian Groffmann, Ania Muntau, Alexandra Tibelius, Eva M. C. Schwaibold, Christian P. Schaaf, Michal Zawada, Lilian Kaufmann, Katrin Hinderhofer, Pamela M. Okun, Urania Kotzaeridou, Georg F. Hoffmann, Daniela Choukair, Markus Bettendorf, Malte Spielmann, Annekatrin Ripke, Martje Pauly, Alexander Münchau, Katja Lohmann, Irina Hüning, Britta Hanker, Tobias Bäumer, Rebecca Herzog, Yorck Hellenbroich, Dominik S. Westphal, Tim Strom, Reka Kovacs, Korbinian M. Riedhammer, Katharina Mayerhanser, Elisabeth Graf, Melanie Brugger, Julia Hoefele, Konrad Oexle, Nazanin Mirza-Schreiber, Riccardo Berutti, Ulrich Schatz, Martin Krenn, Christine Makowski, Heike Weigand, Sebastian Schröder, Meino Rohlfs, Vill Katharina, Fabian Hauck, Ingo Borggraefe, Wolfgang Müller-Felber, Ingo Kurth, Miriam Elbracht, Cordula Knopp, Matthias Begemann, Florian Kraft, Johannes R. Lemke, Julia Hentschel, Konrad Platzer, Vincent Strehlow, Rami Abou Jamra, Martin Kehrer, German Demidov, Stefanie Beck-Wödl, Holm Graessner, Marc Sturm, Lena Zeltner, Ludger J. Schöls, Janine Magg, Andrea Bevot, Christiane Kehrer, Nadja Kaiser, Denise Horn, Annette Grüters-Kieslich, Christoph Klein, Stefan Mundlos, Markus Nöthen, Olaf Riess, Thomas Meitinger, Heiko Krude, Peter M. Krawitz, Tobias Haack, Nadja Ehmke, Matias Wagner Next-generation phenotyping integrated in a national framework for patients with ultra-rare disorders improves genetic diagnostics and yields new molecular findings Preprint, medRxiv 2023.04.19.23288824; doi: https://doi.org/10.1101/2023.04.19.23288824.
Hsieh, TC., Bar-Haim, A., Moosa, S. et al., GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat Genet 54, 349–357 (2022). https://doi.org/10.1038/s41588-021-01010-x
Chengyao Peng, Simon Dieck, Alexander Schmid, Ashar Ahmad, Alexej Knaus, Maren Wenzel, Laura Mehnert, Birgit Zirn, Tobias Haack, Stephan Ossowski, Matias Wagner, Theresa Brunet, Nadja Ehmke, Magdalena Danyel, Stanislav Rosnev, Tom Kamphans, Guy Nadav, Nicole Fleischer, Holger Fröhlich, Peter Krawitz, CADA: phenotype-driven gene prioritization based on a case-enriched knowledge graph, NAR Genomics and Bioinformatics, Volume 3, Issue 3, September 2021. https://doi.org/10.1093/nargab/lqab078Holtgrewe, M.; Stolpe, O.; Nieminen, M.; Mundlos, S.; Knaus, A.; Kornak, U.; Seelow, D.; Segebrecht, L.; Spielmann, M.; Fischer-Zirnsak, B.; Boschann, F.; Scholl, U.; Ehmke, N.; Beule, D. VarFish: Comprehensive DNA Variant Analysis for Diagnostics and Research. Nucleic Acids Research 2020, gkaa241. https://doi.org/10.1093/nar/gkaa241.
Tzung-Chien Hsieh, Martin Atta Mensah et al., PEDIA: Prioritization of Exome Data by Image Analysis, Genetics in Medicine, June 5, 2019. https://doi.org/10.1038/s41436-019-0566-2
Commercial entities interested in software applications and customization of pipelines please contact info@gene-talk.de